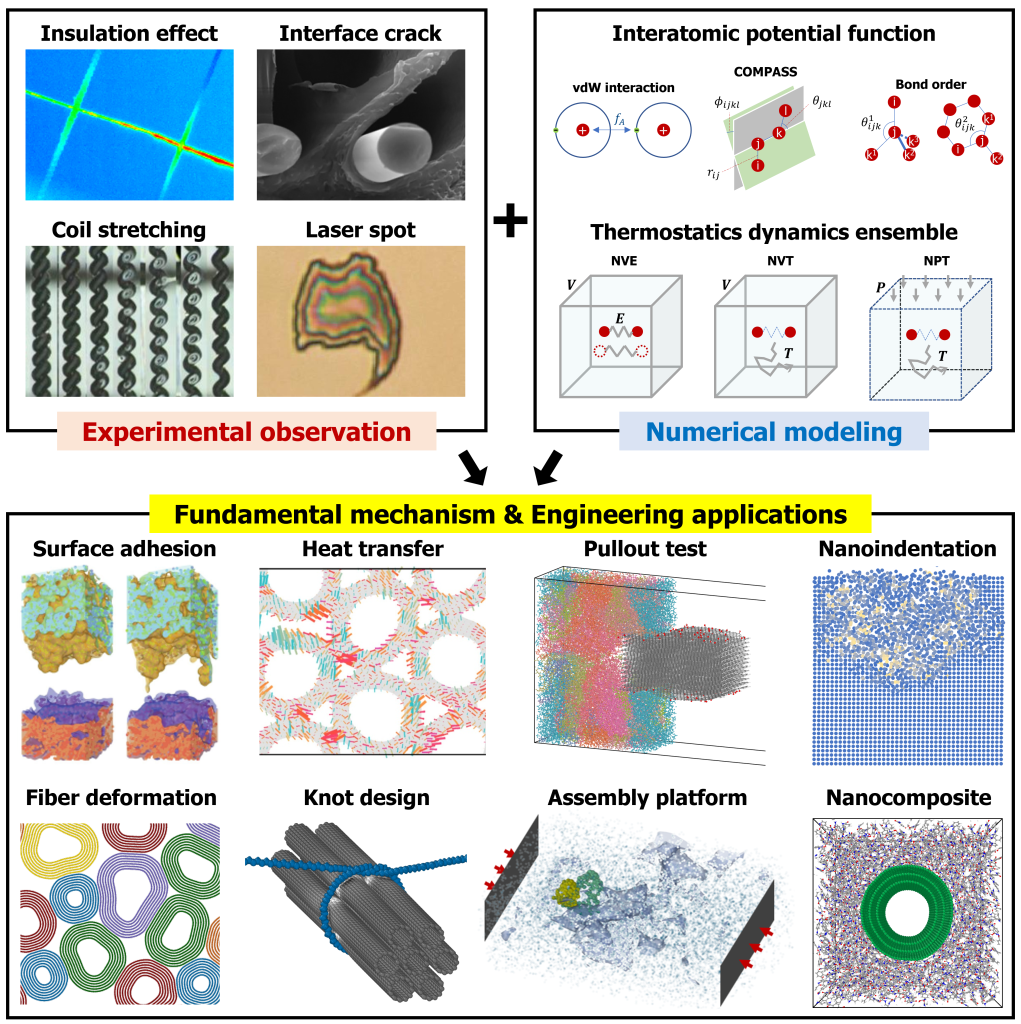
Molecular dynamics (MD) simulations are a powerful tool for validating experimental observations in mechanical engineering by providing atomic-scale insights into material behavior. Many experimental techniques capture macroscopic properties but lack the resolution to reveal underlying molecular mechanisms. MD simulations bridge this gap by modeling atomic interactions based on fundamental physical laws, enabling the study of deformation, heat transfer, and fracture at the nanoscale. By incorporating realistic conditions, MD helps compare computed properties—such as stress distribution or thermal conductivity—with experimental data, improving result interpretation.
In structural materials and tribology, MD simulations are particularly useful for studying wear, friction, and mechanical failure at small scales. They validate experimental techniques like nanoindentation and atomic force microscopy by offering molecular-level explanations for observed behavior. Additionally, MD simulations refine theoretical models and predict material responses under extreme conditions, such as high strain rates or elevated temperatures. By integrating MD with experiments, researchers develop a more comprehensive understanding of mechanical systems, ensuring consistency between theoretical predictions and empirical findings while guiding the design of advanced materials and mechanical components.
